
治療介入による進化(固形癌)!?
がん医療最新論文フォーラム › がん医療最新論文フォーラム › がん医療最新・重要論文 › 治療介入による進化(固形癌)!?
治療介入による進化(固形癌)!?
- このトピックは空です。
-
投稿者投稿
-
-
okazaki yoshihisaゲスト
Nature Medicine
Published: 22 April 2019
『Immunogenic neoantigens derived from gene fusions stimulate T cell responses』
https://pubmed.ncbi.nlm.nih.gov/31011208/
(key word)
融合遺伝子、ネオアンチゲン、ガン免疫療法
(背景)
融合遺伝子が、ネオアンチゲンとなり免疫チェックポイント阻害治療(ICT)が著効した症例の解析結果です。
症例は、転移病巣を伴った頭頸部癌患者さんです。治療前の腫瘍浸潤免疫細胞、腫瘍細胞も遺伝子変異などは少ないにもかかわらずICTにより完全寛解に至りました。
ICTに対する反応性は、腫瘍遺伝子変異量(TMB)が多く、変異によるネオアンチゲンクローン性が高い程、良好であると考えられています。ウイルス抗原、生殖細胞系列抗原も、ICTに対する反応性を決める因子であると考えらています。
TMBが少ないタイプの腫瘍の免疫原性は、融合遺伝子からなるクローン性ドライバー遺伝子変異によって特徴づけられると考えられています。
しかしながら、この融合遺伝子が、TMBが少ない腫瘍のICTに対する反応性とどのような関係があるのかは明らかになっていません。
(内容)
今回症例の腫瘍:=MSK-NH1腫瘍は、肺転移を伴い、頭蓋底原発の頭頸部癌(扁平上皮)(HNSCC)です。
原発巣は切除不能でした。
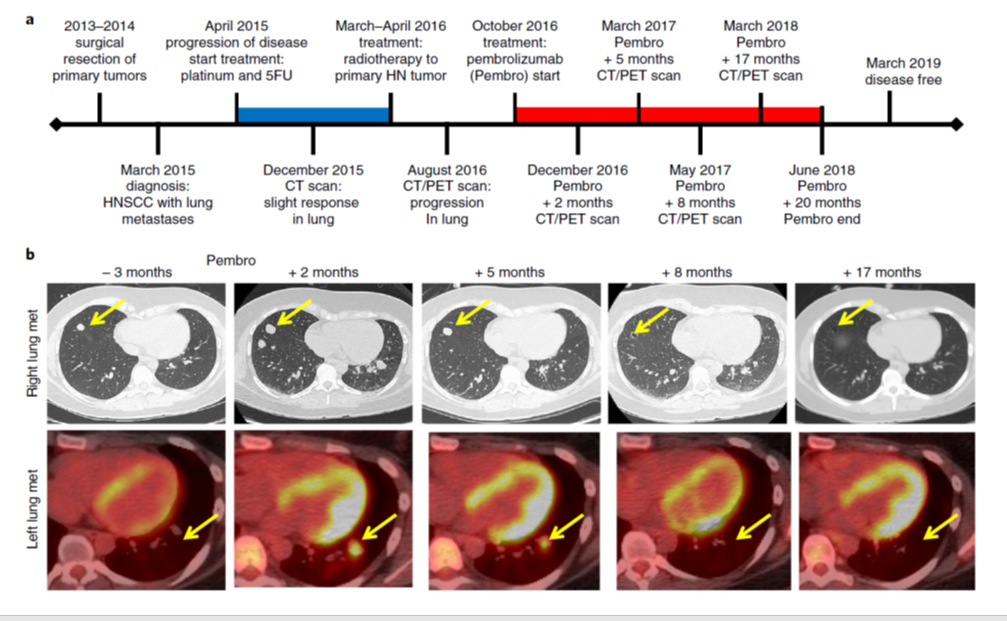
診断後、患者は、プラチナ製剤+5-FU化学療法を施行され、1年間、病状は安定していましたが、その後、再発を認めました(Fig. 1a)。
最終化学療法から6か月後から、抗PD-1抗体 (pembrolizumab)の投与を開始しました。投与開始5ケ月後に画像上の改善を認め、8月ケ後までには、病変の完全消失を認めています(Fig. 1b)。その後、1年間治療を継続し20ケ月後に治療を終了、その後、30ケ月後まで再発なく経過しています。
Fig1.a.b↓
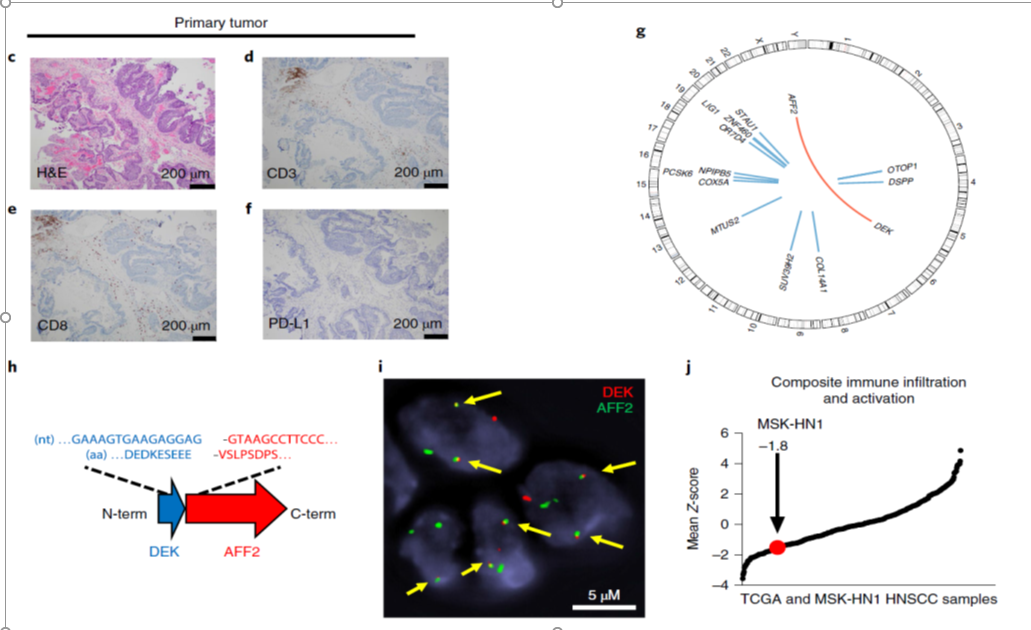
治療開始前の原発巣の組織診断ですが、扁平上皮癌(SCC)で(Fig. 1c)、ヒトパピローマウイルス(HPV)血清型は、調べた範囲で全て陰性でした。
腫瘍微小環境(TME)には、免疫系細胞の浸潤が少なく、多くはCD3⁺CD8⁺T細胞でした(Fig. 1d,e)。
原発巣、肺転移巣ともPDL-1は陰性でした(Fig. 1f)。
免疫療法開始前に原発巣から得られた凍結標本のDNAを全ゲノム解析しました。
DNAの変異によってアミノ酸が置換されるnonsynonymous変異の割合は低かったです(0.47 変異/メガベース;14single nucleotide variations (SNVs) /12 遺伝子)。
注目すべきことに、融合遺伝子DEK–AFF2が見られました(Fig. 1g,h)。
DEKはHNSCCなどのガン種で発癌に関与し、DEK–NUP214融合遺伝子は、急性骨髄性白血病で血液前駆細胞を形質転換させることが知られています。
融合遺伝子DEK–AFF2 は、この症例では、発癌ドライバー遺伝子になっていると考えられました。
何故なら、他の変異遺伝子は、COSMIC cancer gene censusに含まれていませんし、腫瘍をシークエンスしたときに繰り返しディテクトされることもなかったからとのことです。
遺伝子コピー数の変化、遺伝子構造多型なども、全ゲノムに渡って見られなかったそうです。
また、FISH法で、融合遺伝子DEK–AFF2の腫瘍組織内での存在(Fig1.i)も確認でき、RNA-seqデータ解析でも存在を確認できています。
MSK-NH1腫瘍における、免疫細胞浸潤の程度は、HNSCCの中では低い方でした(Fig1.j)。
Fig1.c~j↓
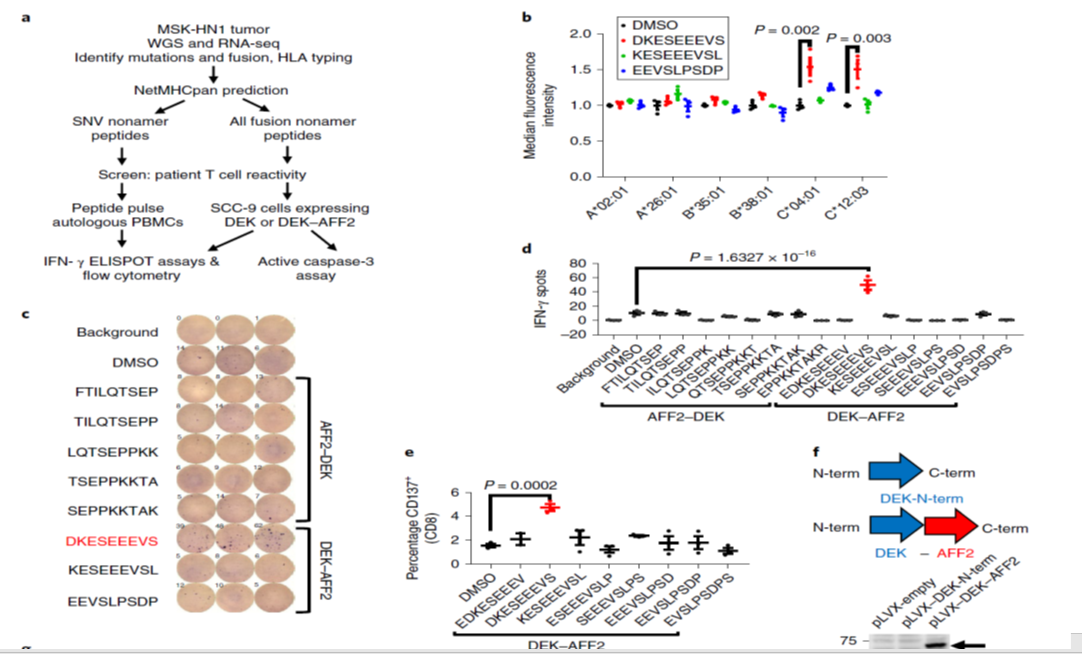
そこで、SNVs や融合遺伝子DEK–AFF2 がT細胞活性化を引き起こすかどうか解析しました(Fig. 2a)。
NetMHCpan 4.0を使って、SNV由来、DEK–AFF2由来の9アミノ酸残基を持ったタンパク質が、この患者さん特異的なHLAs(A*02:01, A*26:01, B*35:01, B*38:01, C*04:01 and C*12:03)に結合するかどうか解析しました。
●HLA-A*02:01:どの変異蛋白質も結合せず(Fig.2a)。
●HLAC*04:01 、HLA-C*12:03 がT2 cells表面に発現した時, DEK–AFF2由来のペプチド (DKESEEEVS)が、これらの分子を安定化させた(P = 0.002, P = 0.003, respectively) (Fig. 2b).
●MSK-NH1腫瘍由来末梢血単核球、DKESEEEVS刺激 T 細胞 存在下で、IFNγの有意な分泌を観察した(P = 1.63 × 10−16) (Fig. 2c,d)。
●CD137⁺の CD8+ T cells の割合も有意に増加した(P = 0.0002) (Fig. 2e)。
●DKESEEEVSで刺激したT細胞活性化は、抗MHC-I 抗体で抑制された。⇔DKESEEEVSアミノ酸の抗原提示は、MHC-Iによることを示唆していた。
●SNVsや他の融合遺伝子では、こうした反応を引き起こすことができなかった(Fig. 2d)。
こうした現象を更に確認するために、
DEK–AFF2遺伝子の融合部分で切断した、
DEK-N-term遺伝子とDEK–AFF2遺伝子をHLA-C*04:01-陽性 SCC-9 細胞に導入・発現させ(pLVX-Puro:レンチウイルスベクター)(Fig. 2f)、T細胞と培養しました
Fig2.a~f↓
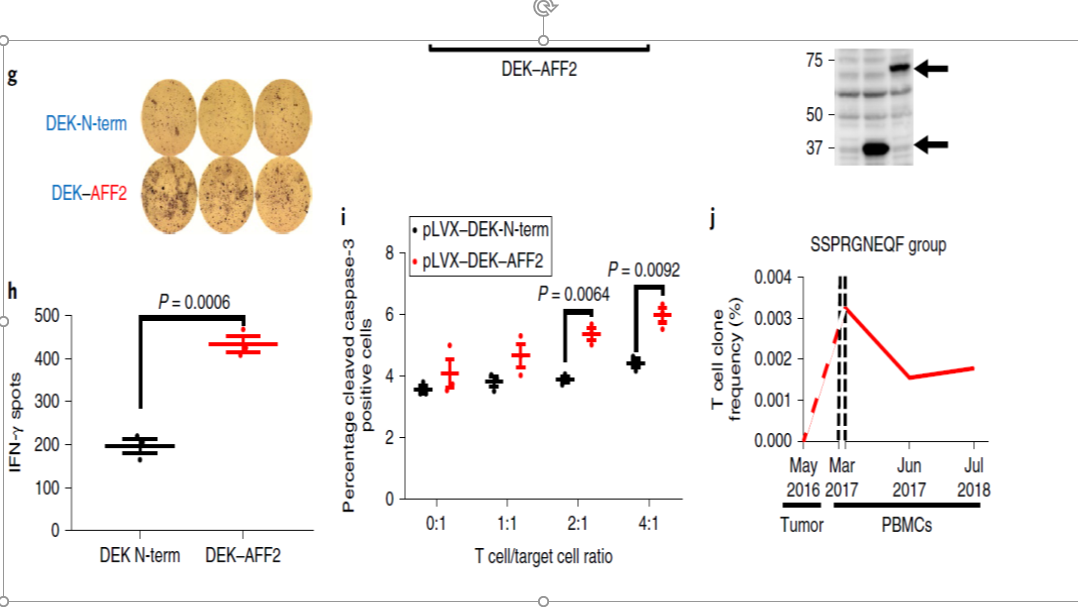
●DEK–AFF2蛋白陽性時に、IFNγの有意な分泌がみられた(P = 0.0006)。
●SCC-9細胞を抗MHC-I 抗体で処理したときにIFNγ分泌は抑制されました(Fig. 2g,h)。
●活性型カスパーゼ3で誘導されるアポトーシスもDEK–AFF2蛋白陽性時に増加しました(P = 0.0064, 0.0092 ) (Fig. 2i)。
同様の現象が、COS-7細胞での実験でも観察されています。
次に、患者の治療前の生検腫瘍組織中T細胞と、治療中の末梢血中T細胞のT細胞受容体(TCR)β鎖のCDR3領域のDNAを解析比較しました。
治療中の末梢血中DKESEEEVSペプチド反応性T細胞のCDR3は、治療前の生検組織と比べ著名に増加していました(P = 0.016)。
DKESEEEVS反応性T 細胞中や患者末梢血中に最も多かったT細胞クローンは、治療開始(2016年10月~pembrolizumab開始)前の生検組織中には存在していませんが、
治療開始後、5か月目(2017年3月)には、末梢血中に出現しはじめ、その後、腫瘍が治療に反応するにつれて減少しまし、治療開始後21ケ月後(2018年7月)も血中に存在していることが確認できました(Fig. 2j)。
Fig2.g~j↓
こうしたデータは、DEK–AFF2 ネオアンチゲン特異的なT 細胞反応は、腫瘍縮小過程で発生することを示唆します。
また、このとは、腫瘍反応性T細胞は、未治療腫瘍内TCRレパトアには見つからず、免疫治療中の腫瘍縮退期に出現するといった、以前からの主張にも合致するようです。
しかしながら、エピジェネティック変異による生殖細胞系列ガン抗原などをネオアンチゲンから除外することができていない点も考察しています。
今回の腫瘍では、27コのよく知られた“ガン生殖細胞系列抗原”の強発現は確認されていないし、WGSで確認された他の構造変異もありませんでした。
また、発生しうる選択的スプライジングからネオエピトープが発生する可能性も検討していますが、そのような候補物は発見できなかったようです。
このようなデータを総合的に判断して、,融合遺伝子DEK–AFF2以外の変異=ウイルス、生殖細胞系列変異、選択的スプライジング変異などは、今回の腫瘍では腫瘍縮小を誘導するネオアンチゲンにはなりえないのではないかと結論しています。
次に、他の似たような融合遺伝子ペプチドが、ネオアンチゲンになっていないか、頭頸部ガンの一種で似たような腫瘍=Adenoid cystic carcinomas (ACCs)で、MYB–NFIB融合遺伝子の役割を同じような方法で解析し、MYB–NFIB融合遺伝子もT細胞刺激能力を有するネオアンチゲンになりうる可能性を指摘しています(原論文のFig3.a~j,Fig4.a~fを参照ください。今回はupしていません)。
最後に、一般的な融合遺伝子は、免疫療法中に腫瘍免疫環境を編集するのか?
異なる腫瘍腫のメラノーマで検討しています。抗PD-1抗体治療を受けた患者さんで、治療前と治療中の生検サンプル(n=42)をRNA-seq解析しました。
臨床経過:CR/PR、SD、PDの3群に分けて解析すると、Fif4.fのように、CR/PR群で治療後に融合遺伝子の数が減少しています。ただし、メラノーマはTMBが高く、今回主に注目したTMBが低いSCCとは事情が異なる可能性もあります。
Fig4.f↓
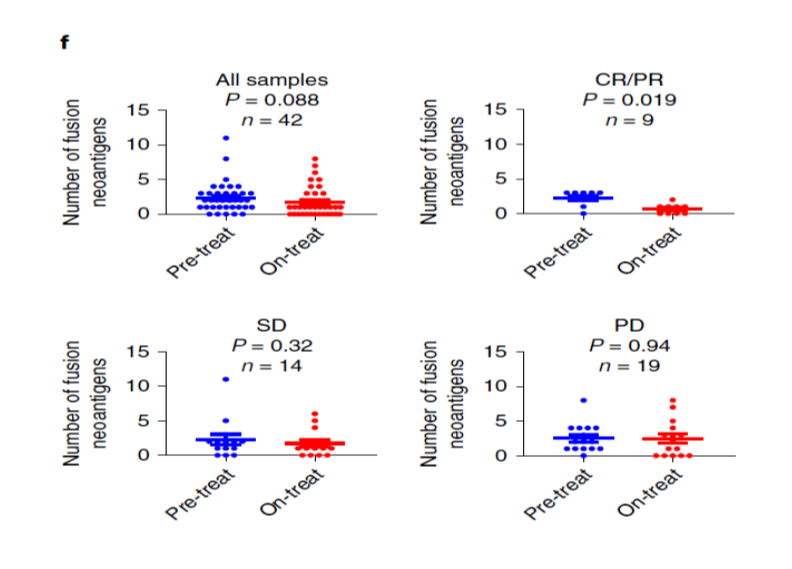
こうした、融合遺伝子は、
ユーイング肉腫のEWS–FLI1融合遺伝子、
肺腺癌の EML4–ALK融合遺伝子,
前立腺癌の TMPRSS2–ERG融合遺伝子、
転移癌などでのETV6–RUNX1, FGFR3–TACC3, TEL–AML1融合遺伝子なども、
有望なネオアンチゲンにる可能性を示唆しているのではないかと結論しています。
(感想)
2019年の論文で“最新”ではありませんが、今回、ネオアンチゲンというより、“ガンの治療進化”の切り口でも面白いかな?と思い読んでみました。
頭頸部扁平上皮癌でTMBが低い腫瘍とのことです。
しかし、免疫チェックポイント阻害薬治療を開始すると、それに呼応するかのように、ガン免疫原性を持ったネオアンチゲン(DEK–AFF2融合遺伝子)が出現し、治療終了後も存在しつづけ、腫瘍の治療反応性維持に関与する可能性が示唆されています。
このように、ガン細胞は、“治療選択圧”によって、治療にプラスの方向にも、治療にマイナスの方向にも、ツギツギに変化していく“動的”な存在である可能性が大きいとの印象を新たにしました。
-
okazaki yoshihisaゲスト
融合遺伝子が、いつ?つまり、免疫療法前のケモラジオに反応して生じたのか?それより前の段階で生じたのかは、今回の論文では、わかりませんね!
実際は、どうなのでしょう?
治療反応性のT細胞は、免疫療法開始後に出現しているようですが?
-
-
投稿者投稿
